For decades, scientists have tried to answer a deceptively simple question: Why are some substances allergens?
The Airway's Living Border
Every breath brings the outside world deep into the body. The lungs face an unusual biological challenge. Their enormous internal surface must remain thin and permeable enough to exchange oxygen and carbon dioxide while simultaneously protecting the body from everything that arrives with a breath.
Along with oxygen, we inhale pollen fragments, fungal spores, bacteria, viruses, smoke particles, animal proteins and thousands of environmental chemicals. Standing between this complex mixture and the immune system is a thin layer of epithelial cells lining the respiratory tract.
These cells were once viewed primarily as a passive barrier. Scientists now understand that the airway epithelium functions more like a sophisticated environmental surveillance system.
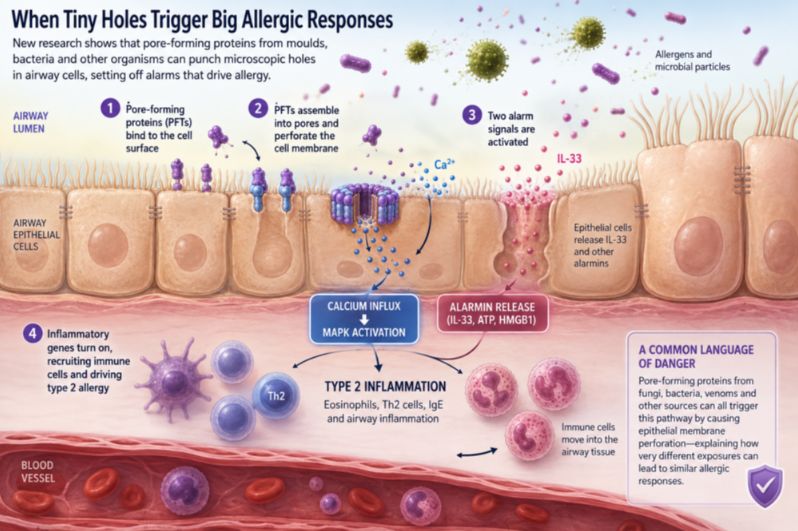
When epithelial cells detect injury or stress, they can release molecular alarm signals, including the cytokine IL-33. These signals alert nearby immune cells and can initiate the cascade that ultimately produces eosinophilic inflammation, mucus secretion, airway hyperresponsiveness and IgE production.
The new study goes a step further by identifying a physical event that may trigger this alarm system: membrane perforation.
A Mold with a Molecular Hole Punch
Alternaria alternata is widespread in the environment and has long been associated with respiratory allergy and asthma exacerbations. Researchers knew that extracts from the mold could provoke strong inflammatory responses, but the mechanisms involved were not fully understood.
One Hole, Two Alarm Systems
A cell membrane is not simply packaging. It maintains the carefully controlled chemical differences between the inside and outside of a cell. Sodium, potassium and calcium ions are kept at specific concentrations. Proteins and signaling molecules are held within particular compartments. These gradients allow cells to communicate, produce energy and respond to their environment.
Punching even a small hole in this system can create biological chaos. The Alternaria pores activated at least two major alarm pathways.
The first involved the release of IL-33. As the epithelial membrane was disrupted, IL-33 entered the extracellular environment, where it could help activate type 2 immune responses. The researchers also detected other danger-associated molecules, including ATP and HMGB1.
The second pathway began when calcium ions flooded through the new pores.
Calcium is best known as a component of bone, but at the cellular level it is also one of biology's most versatile messengers. Cells normally maintain very low concentrations of free calcium in their interior compared with the surrounding environment. When calcium suddenly enters the cell, it can rapidly alter cellular behavior.
In the experiments, calcium influx activated a signaling network called the MAPK pathway, involving proteins including MEK and ERK. This pathway switched on inflammatory genes that help recruit and organize immune cells in the airway.
The two alarm systems were related but not identical. Even in mice lacking IL-33, pore formation could still activate MAPK signaling and inflammatory gene expression. The implication is that membrane injury does not pull a single fire alarm. It activates several.
The Cell Does Not Have to Die
Perhaps the most consequential finding was that epithelial cells did not have to be destroyed for inflammation to begin. At relatively low concentrations, the pore-forming proteins could alter calcium signaling and activate inflammatory pathways without causing immediate cell death.
That distinction may be important for understanding chronic airway disease. We tend to imagine tissue injury as something dramatic: a burn, a wound, a toxic exposure that kills cells outright. But biology is filled with subtler forms of stress. A cell may remain alive while its membrane is temporarily disturbed. It may repair itself after injury. Yet during that period, it can release alarm signals and change the behavior of neighboring cells.
This raises the possibility that some allergic inflammation may emerge not from catastrophic epithelial destruction but from repeated episodes of small-scale cellular disturbance.
A pore opens. Calcium enters. An alarmin escapes. Inflammatory genes switch on. Immune cells arrive. Then the process happens again. Over time, repeated environmental encounters could potentially reinforce an inflammatory state in a susceptible airway.
The study does not prove that this sequence explains all asthma or allergy. Human allergic disease is far more complicated, involving genetics, viral infections, pollution, microbiome changes and many other influences. But it offers a plausible mechanism by which repeated environmental exposure could be translated into immune activation.
When the Scientists Disarmed the Mold
Discovering that two mold proteins can trigger inflammation does not prove that they are responsible for the mold's broader allergenic effects.
To test that possibility, the researchers genetically engineered Alternaria strains unable to produce either Aeg-S or Aeg-L.
The results were dramatic. Extracts from the altered mold strains largely lost their ability to trigger IL-33 release and MAPK activation. In experimental animals, they also failed to produce the same accumulation of eosinophils and TH2 cells seen with ordinary Alternaria extract.
The researchers then restored the missing proteins. The inflammatory activity returned.
These experiments provided strong evidence that the pore-forming machinery was not merely an incidental feature of the mold. In the experimental models, it was a crucial driver of its ability to provoke allergic airway inflammation.
How Injury Can Teach the Immune System What to Fear
The pore-forming proteins did something else that may be equally important: they helped the immune system become allergic to other proteins.
When researchers administered Aeg-S and Aeg-L together with experimental antigens, the animals developed antigen-specific TH2 cells and IgE antibodies. In effect, the membrane injury acted as an immune amplifier. Immunologists call such substances adjuvants. Vaccines often use adjuvants to tell the immune system that an accompanying antigen deserves attention. But different danger signals can shape different kinds of immunity.
The Alternaria proteins did not simply produce generic inflammation. They favored the IL-4- and IL-13-associated immune pattern characteristic of type 2 responses. This suggests a provocative scenario.
Imagine that the airway simultaneously encounters a harmless environmental protein and a molecule that damages epithelial membranes. The immune system receives both pieces of information at once: here is a foreign protein, and here is tissue danger.
Under the right circumstances, the immune system may connect them. The harmless protein can become something to remember, and something to attack during future exposures.
Different Threats, the Same Cellular Injury
The researchers then broadened their investigation beyond Alternaria. They tested pore-forming proteins from a remarkably diverse collection of organisms: another mold, a bacterium, a sea anemone, an earthworm, a mushroom and other fungi.
These proteins were not identical. Some recognized cholesterol in cell membranes. Others targeted sphingomyelin, other lipids or carbohydrates. Their molecular architectures varied considerably. Yet all could provoke allergic airway inflammation in the experimental models.
This may help explain one of allergy's enduring mysteries. Molds, venoms and microorganisms are biologically different. The immune system may not need a universal receptor capable of recognizing every allergenic substance. Instead, it may recognize the shared damage those substances cause.
The molecular weapons are different. The cellular consequence is similar. A hole has appeared where a hole should not be.
Why Evolution Might Have Built Such an Alarm
From an evolutionary perspective, detecting membrane perforation makes sense. Many dangerous organisms produce molecules that damage cell membranes. Parasites, fungi, bacteria and venomous animals have independently evolved proteins capable of disrupting the cellular boundary.
An animal that could rapidly detect such damage would have an advantage.
Type 2 immunity is associated not only with allergy but also with tissue repair, mucus production, barrier restoration and defense against certain parasites and toxins. Seen in this light, the allergic response may represent the inappropriate activation, or chronic overactivation, of an ancient protective system.
A pore in a membrane is a reliable sign that something has gone wrong. The difficulty is that a defense system designed to respond to danger can become costly when repeatedly activated in modern environments or in biologically susceptible individuals.
The same pathways that may once have helped expel parasites or repair toxin-damaged tissue can contribute to chronic mucus production, airway hyperresponsiveness and eosinophilic inflammation. Protection and disease may arise from the same machinery.
Could Allergy Be Interrupted Earlier?
Most treatments for allergic disease intervene after the inflammatory process is already underway. Antihistamines block the effects of histamine. Corticosteroids suppress broad inflammatory programs. Modern biologic drugs can target IgE or specific cytokine pathways, including IL-4, IL-5 and IL-13 signaling.
The new research raises a different possibility: Could inflammation be interrupted closer to its epithelial origin? Theoretically, the steps between membrane perforation, calcium influx and MAPK activation could offer therapeutic targets. Preventing the pore from forming, accelerating membrane repair or selectively modifying the epithelial response to calcium influx might stop the cascade before large numbers of immune cells are recruited.
That prospect remains speculative.
Calcium and MAPK signaling are fundamental to normal cell biology, and indiscriminate inhibition could cause serious unwanted effects. Any future therapy would need to act with great precision. But the conceptual shift may prove more important than any immediate drug target.
The findings encourage researchers to look upstream, to the earliest seconds and minutes after an environmental exposure meets the epithelial surface.
The Body May Recognize Damage before Identity
For much of modern immunology, scientists have focused on recognition: Which receptor recognizes which molecule? Which antibody binds which antigen? Which immune cell responds to which cytokine?
Those questions remain essential. But the Alternaria study points toward another dimension of immune surveillance. Sometimes the body may first ask not What is this? but What has this done to me?
The airway epithelium sits at that intersection. It is simultaneously a physical barrier, an environmental sensor and an immune signaling network. It experiences the outside world before deeper tissues do. When its membranes are disrupted, it can transform a microscopic physical injury into a body-wide biological message.
The study by Shi and colleagues suggests that the beginning of an allergic response may sometimes be extraordinarily small: a molecular complex assembling on a cell surface, a channel opening across a membrane and a sudden rush of calcium into the cell.
From that tiny event, a much larger response can follow. An alarmin is released. Inflammatory genes turn on. Eosinophils arrive. TH2 cells accumulate. IgE antibodies appear. The immune system remembers.
A microscopic hole may become the first chapter of an allergic disease.
Reference
1. Shi K, Lv Y, Zhao C, et al. Epithelial cell membrane perforation induces allergic airway inflammation. Nature. 2025;645(8080):475-483. doi:10.1038/s41586-025-09331-1
2. Whetstone CE, Ranjbar M, Omer H, Cusack RP, Gauvreau GM. The Role of Airway Epithelial Cell Alarmins in Asthma. Cells. 2022;11(7):1105. Published 2022 Mar 24. doi:10.3390/cells11071105
3. Duchesne M, Okoye I, Lacy P. Epithelial cell alarmin cytokines: Frontline mediators of the asthma inflammatory response. Front Immunol. 2022;13:975914. Published 2022 Oct 14. doi:10.3389/fimmu.2022.975914
4. Snelgrove RJ, Gregory LG, Peiró T, et al. Alternaria-derived serine protease activity drives IL-33-mediated asthma exacerbations. J Allergy Clin Immunol. 2014;134(3):583-592.e6. doi:10.1016/j.jaci.2014.02.002
5. Hristova M, Habibovic A, Veith C, et al. Airway epithelial dual oxidase 1 mediates allergen-induced IL-33 secretion and activation of type 2 immune responses. J Allergy Clin Immunol. 2016;137(5):1545-1556.e11. doi:10.1016/j.jaci.2015.10.003